
Порушення в кількості чи структури хромосом призводять до вроджених генетичних захворювань (синдром Дауна, Свайера, Клайнфельтера та ін). Такі хвороби не піддаються лікуванню, тому що це хромосомні патології, терапевтичні методи спрямовані на усунення симптомів і підтримання гідного рівня життя. До одним з рідкісних генетичних захворювань відноситься синдром «котячого крику» (СКК).

Що таке синдром «котячого крику»?
Вперше синдром Лежена був описаний в 1963 році французьким лікарем-педіатром, видатним генетиком, який присвятив свою кар’єру вивчення хромосомних аномалій, Жеромом Леженом. Дана мутація має й інші назви, наприклад, синдром делеції хромосоми 5р-, причини виникнення патології. Інша назва — синдром «котячого крику» — аномалія отримала з-за характерного звуку, видаваного хворими дітьми.
Синдром Лежена — досить рідкісне генетичне порушення, яке буває у 1 новонародженого з 50 000. Найчастіше зустрічається у дівчаток, співвідношення хворих жіночої і чоловічої статі — 4 до 3.
Причини виникнення синдрому Лежена у дітей
Людина має 22 пари хромосом і 23-ю статеву пару ХХ або ХУ, яка визначає його статеву приналежність. Кожна хромосома має довге плече, яке позначається літерою q, і короткий р. Плечі поділяються на пронумеровані ділянки, кожен з них визначає місцеположення тисяч генів.
СКК зумовлений частковою моносомией 5 хромосоми. В результаті хромосомних перебудов — делецій — відбувається втрата деякої ділянки хромосоми. При аномалії Лежена мутації зачіпають коротке плече і найчастіше 14 ділянку, тому хвороба часто позначається як 5р14.
Клітини, що містять хромосомні патології, не можуть продукувати необхідні хімічні речовини. Це породжує дефектні білки, які викликають симптоми хвороби.

При СКК можливі різні варіанти мутацій:
- повна відсутність плеча — призводить до важких патологій, часто несумісним з життям;
- втрата частини плеча — зустрічається найчастіше, симптоми залежать від загубленого фрагмента;
- кільцева хромосома — обидва коротких плеча замикаються і з’єднуються між собою, при цьому відбувається делеція теломер.
Найбільш легка форма аномалії зустрічається при мозаицизме. Спочатку зародок отримує нормальні хромосоми. Мутації починаються після утворення зиготи. З-за цього більшість клітин має нормальний хромосомний набір, а меншість — аномальний.
Чому виникає така патологія, науковцям невідомо. Тільки 10% випадків пов’язані з спадковістю, причому в 80% з них спадкування йде від батька. Інші випадки викликані спонтанними мутаціями.
Передбачувані фактори, які збільшують ризик СКК:
- радіаційне випромінювання;
- вірусні ураження матері під час вагітності;
- вплив хімікатів і токсичних речовин;
- вплив антибіотиків, препаратів для хіміотерапії та інших медикаментів;
- вживання психоактивних речовин;
- вік матері більше 40 років.
Супутні симптоми

Тяжкість симптомів варіюється в залежності від того, яку ділянку хромосоми піддався делеції. Для дітей з аномалією характерні наступні ознаки:
- Специфічний плач, який нагадує котяче нявкання. Саме за цією ознакою синдром отримав свою назву. Причина такого плачу полягає в зміні будови гортані — вона звужена, надгортанник зменшений, на слизовій оболонці присутні складки, хрящі м’які. Як правило, цей симптом зникає в 1-2 роки. Послухати, як звучить характерний плач, можна на відео.
- Відставання у фізичному розвитку. Немовлята народжуються з масою тіла, яка на 20% меншою, ніж здорові діти. У них знижений тонус м’язів. Голова і головний мозок занадто маленькі порівняно з іншими частинами тіла, що супроводжується розумовою відсталістю різного ступеня вираженості.
- Особа — місяцеподібне, очі широко розставлені. Присутній епікантус — шкірна складка біля внутрішнього куточка ока. З віком особа витягується, стає довгим і вузьким. Як виглядає дитина з СКК, можна побачити на фото.
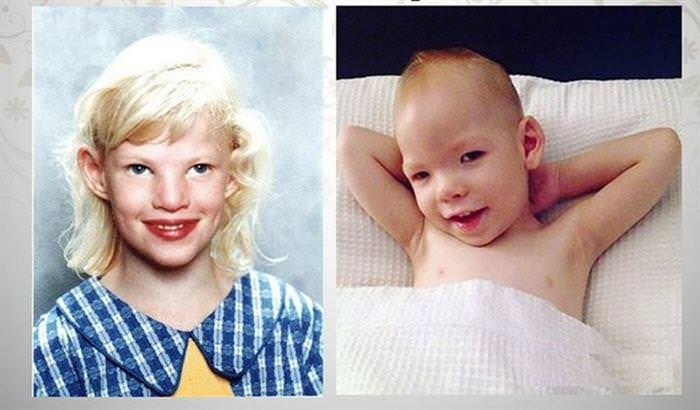
- Неправильне будова вуха. Вони надто маленькі, мають шкірні складки.
- Пороки внутрішніх органів. Найчастіше зустрічається патентний артеріальна протока, при якому протока між легеневою артерією і аортою не закритий. Іноді у немовлят буває мальротация кишечника.
- Відставання в розумовому розвитку. Залежно від ступеня тяжкості патології розвиваються помірна або сильна розумова відсталість, затримка мовлення. Всього 50% дітей вміють користуватися ложкою до 3,5 років.

Як діагностують захворювання?
Якщо у батьків в роду зустрічалися захворювання, викликані хромосомними аномаліями, то лікарі рекомендують на етапі планування вагітності на ранніх термінах гестації пройти ряд генетичних досліджень:
Тактика лікування
Оскільки хвороба «котячого крику» — це генетичне захворювання, то усунути його точні причини і вилікувати неможливо. Тактика лікування спрямована на усунення і мінімізації симптомів.
Дитині з ранніх років показано реабілітаційні заняття, з допомогою яких можна впоратися з затримкою фізичного і розумового розвитку. Зазвичай вони включають в себе фізіотерапію, роботу з дефектологом, психологом, логопедом.
При серцевих вадах клапанів, розщеплення піднебіння, неправильно сформованих вушних раковинах, століттях малюкові роблять операції, спрямовані на виправлення цих наслідків СКК. Протягом усього життя пацієнт повинен спостерігатися у терапевта, кардіолога, невролога, хірурга і психотерапевта.

Прогноз і можливі ускладнення
Хвороба не позначається на тривалості життя. При належному догляді пацієнти доживають до похилого віку. Найдавнішому з відомих пацієнтів був 61 рік. Залежно від тяжкості захворювання людини можна адаптувати до соціального життя, поліпшити психомоторні навички та мова, він може навіть отримати професію. Якщо ж хвороба приймає важку форму, то хворому потрібно постійний опікун.
СКК не впливає на фертильність. Чоловіки і жінки можуть зачати дитину, який буде здоровий. Однак у 50% випадків синдром Лежена проявляється і у дітей.
У 10% випадків відхилення бувають несумісні з життям. Такі діти вмирають у перший рік після появи на світло. Частіше всього служать причиною смерті вади серця, аспіраційна пневмонія, респіраторний стрес. Якщо дитина не отримує належного догляду та медичної допомоги, його тривалість життя рідко перевищує 10 років.

Чи існують заходи профілактики виникнення у дитини синдрому Лежена?
Оскільки причини, які викликають спонтанні мутації в хромосомах, невідомі, то профілактичних заходів, які дозволили б на 100% уникнути виникнення патології, не існує. Мінімізувати ризики народження хворої дитини допоможуть здоровий спосіб життя батьків, проживання в екологічно сприятливому середовищі, всебічне обстеження перед зачаттям. Незважаючи на те, що такі заходи є недоведеним способом профілактики СКК, вони в цілому вбережуть майбутньої дитини від інших недуг.
Перед плануванням вагітності майбутнім батькам, які мають родичів з генетичними аномаліями або входять у групу ризику, слід здати кров на генетичний аналіз. При народженні дитини з СКК батьки зобов’язані пройти обстеження. Якщо один з них є носієм структурної перегрупування, то другий дитина швидше за все теж буде хворий. Оскільки мутації в більшості випадків є спонтанними, то ймовірність рецидиву складає менше 1%.
