
У давнину генетичні аномалії вважалися проявами містичного впливу. З початку 20 століття до наших днів завдяки розвитку генетики медицина ступнула далеко вперед. Вивчено причини появи багатьох захворювань, у тому числі викликаних генними мутаціями. Порушення хромосомної структури передаються у спадок. Такі захворювання не лікуються, наприклад, синдром Шерешевського-Тернера. В яких формах проявляється захворювання? Як діагностувати і лікувати симптоми?
Поняття синдрому Шерешевського-Тернера
Вперше аномалія була описана в 1926 році Миколою Шерешевским. Ендокринолог Тернер через кілька років відкоригував характер захворювання, довівши залежність вродженого гіпогонадизму з статевим інфантилізмом, суглобовими деформаціями, зовнішніми проявами.
Брак хромосоми Х викликає порушення розвитку і функціонування статевих залоз. Хворі мають недорозвинені статеві органи, вторинні статеві ознаки, маленький зріст, патології внутрішніх органів.
Новонароджені часто з’являються на світ раніше терміну. У них можуть повністю відсутні яєчники. З віком спостерігається відхилення статевого, моторного розвитку.
Це найпоширеніша хромосомна патологія у жінок. Медичне назва – Синдром Тернера, код за МКХ-10 Q96. Частота зустрічальності – один випадок зустрічається на 4000 новонароджених. Нерідко відбувається викидень на ранніх термінах. Ці факти статистикою не враховуються.
Основні симптоми та неспецифічні ознаки

При пологах у строк малюк народжується з маленьким зростом до 48 см, вагою до 2,9 кг Новонароджений може за іншим фізіологічним ознаками не відрізнятися від здорових дітей або мати явні патологічні зміни зовнішності. Хромосомну моносомию можна запідозрити при вагітності, коли жінка відчуває сильний токсикоз. Виношування супроводжується постійною загрозою переривання. Діти майже завжди народжуються недоношеними.
Ознаки синдрому Шерешевського-Тернера у ранньому віці:
- бічні складки на короткій шиї;
- набряклість ніг, рук;
- вроджений порок серця;
- лімфостаз;
- патології сечовидільної системи;
- ускладнена міміка з-за вродженої слабкості м’язів обличчя;
- у внутрішньому куті ока утворюється шкірна складка – эпикант;
- часте утворення родимок;
- зригування після їжі;
- неспокійна моторика;
- слабка сосательная функція.

Основний симптом хромосомної патології Тернера – статевий інфантилізм, інакше гіпогонадизм первинного походження. У пацієнток недорозвинені зовнішні і внутрішні статеві органи, діагностується гіпоплазія матки. Замість яєчників розвиваються двосторонні фіброзні тяжі, у яких немає фолікул. Менструації відсутні. Більшість пацієнток безплідні.
У міру дорослішання у дітей спостерігається відставання в мовленнєвому, а також фізичному розвитку. Може виникнути кондуктивна приглухуватість, яка пояснюється частими отитами. Уражаються кістки, суглоби, що виражається дисплазією кульшових суглобів, сколіозом, вальгусним викривленням ліктів. У хворих рано розвивається остеопороз через нестачу естрогенів. Це обумовлює часті переломи кісток.
Дорослі пацієнти мають невисокий зріст – до 140-145 див. Їм характерні такі особливості зовнішності: широка грудна клітка, як бочка, коротка шия, складки у вигляді крил по боках шиї, неправильний прикус, раковини деформовані вуха. Як виглядає жінка з генетичною патологією – показано на фото. Страждає зір. Часто розвивається міопія, косоокість, дальтонізм.
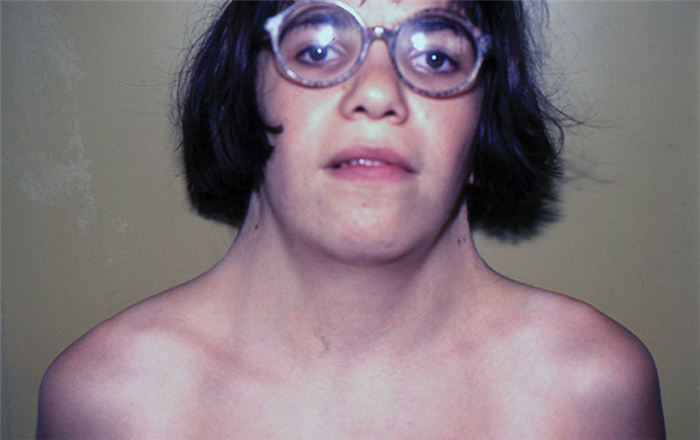
Порушення розумового розвитку зазвичай не спостерігається. Іноді діагностується олігофренія. Генетична аномалія часто супроводжується такими захворюваннями, як цукровий діабет 1, 2 типу, вітіліго, гіпотиреоз (нестача гормонів щитовидної залози), ожиріння, пігментні невуси. Високий ризик онкологічних утворень в прямій кишці.
У деяких пацієнтів виявляються неспецифічні додаткові ознаки: велика відстань між сосками, підвищена пігментація шкіри. Ендокринний дисбаланс викликає накопичення пігменту, що проявляється у вигляді родимих плям, родимок.
Причини виникнення синдрому
Вся інформація про людину, його предків закодована в 46 хромосомах. Код несуть в собі знаходяться в певному порядку гени. 44 хромосоми передають загальну інформацію про організмі. Дві визначають стать. У жінок дві хромосоми Х, у чоловіків – ХУ. Коли в жіночому генетичному коді відсутня одна хромосома Х, це є причиною синдрому Тернера-Шерешевського.
Недолік статевої хромосоми з’являється в результаті відхилення каріотипу від норми. Передбачити патологію неможливо. Хвора дитина може народитися у здорових батьків.
Вивчення спадкової аномалії виявило зв’язок її виникнення з наступними факторами:
- перенесені матір’ю статеві інфекції;
- несприятлива екологія;
- електромагнітне випромінювання;
- виснаження організму матері;
- тривале голодування перед зачаттям;
- зловживання алкоголем при виношуванні малюка.
Форми захворювання і тип успадкування
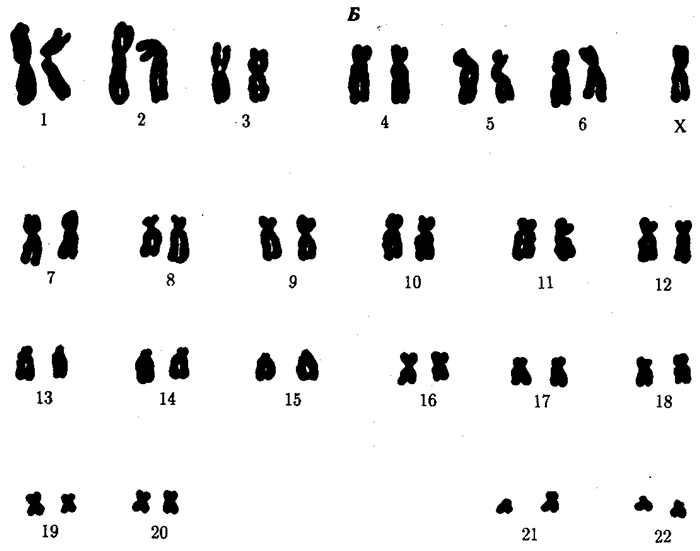
Генетичний синдром класифікують на три різновиди, виходячи з каріотипу пацієнта:
До кінця не вивчено, як відбувається спадкування синдрому. Залежність появи у потомства короткої хромосоми з віком, станом здоров’я батьків не виявлено.
Діагностичні заходи
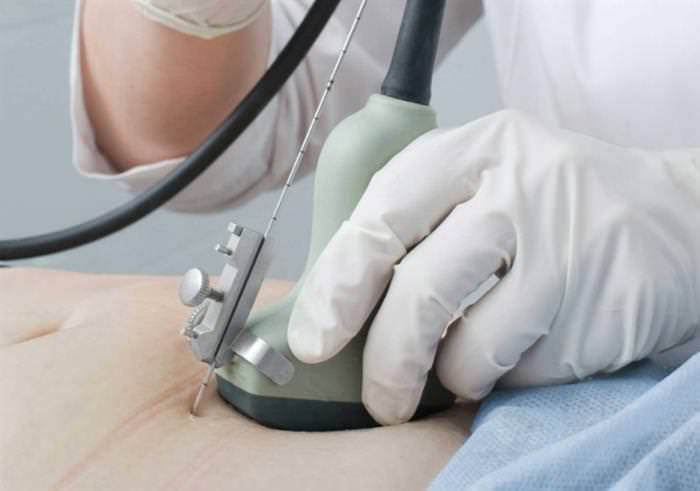
Виявлення синдрому в період вагітності відбувається рідко. На плановому ультразвуковому дослідженні розглянути складки на шиї плода практично неможливо. Перинатальні методи дослідження робляться за суворими показаннями, так як можуть бути небезпечні для плода. При підозрі на розвиток синдрому Шерешевського-Тернера застосовують способи інвазивної діагностики. Проводяться генетичні аналізи, біопсія хоріона, кордоцентез, амніоцентез.
Після появи на світ новонародженого первинний діагноз встановлюється, якщо при огляді виявлені характерні зовнішні прояви. Підтвердити або спростувати діагноз допомагає каріотипування. Генетики проводять вивчення кожної хромосоми з описом специфічних ознак. Розглядається форма, будова, параметри хромосомної структури. Для цитогенетичного аналізу береться кров з вени. Виділяються кров’яні лімфоцити в стадії мітозу. Лікар-генетик реагентами зупиняє процес поділу, потім аналізує хромосомний ряд.
Додаткові дослідження проводяться з метою диференціювання патології від деяких інших зі схожими симптомами. Схожі психоневрологічні, фізіологічні ознаки спостерігаються у хворих з синдромами Нунана, Рокитанского-Кюстнера-Майера. Додаткові методи постановки діагнозу:
- УЗД внутрішніх органів;
- кардіограма для вивчення роботи серця;
- загальний аналіз крові, сечі;
- визначення статевого хроматину;
- аналіз на гормони;
- рентген кісток стоп, кистей;
- МРТ, КТ.
Генетик проводить опитування батьків для виявлення факторів, які можуть вплинути на виникнення синдрому: алкоголізм, виснаження. З’ясовується, чи були в роду якісь генетичні захворювання до четвертого коліна. Крім генетика, потрібно огляд профільних фахівців.
Основне лікування і додаткові методи терапії
Як і інші генетичні патології, синдром Тернера-Шерешевського не можна вилікувати повністю. Терапія допомагає відкоригувати гормональний дисбаланс, стимулювати зростання, дає можливість пацієнтам жити повноцінним життям.
Мета полягає в регуляції статевих функцій, корекції зовнішніх проявів, нормалізації менструацій, відновлення репродуктивної функції.
Лікування різнопланове. Дітям призначається лікувальний масаж, комплекси вітамінів, курси ЛФК. З 13 років починається замісна терапія естрогенами. Це стимулює статеве дозрівання – розвиваються вторинні статеві ознаки, збільшуються розміри матки, статура приймає жіночні обриси, купірується розвиток остеопорозу. Естроген слід приймати до клімаксу.
Період активного росту дитини до 15 років вимагає гормональної терапії ін’єкціями. Це необхідно для стимуляції росту. Застосовуються препарати Генотропин, Соматотропін в поєднанні з прогестероном.
Для успішного лікування важливо здорове, повноцінне харчування, раціональне співвідношення роботи та відпочинку. Добре оздоровлюють фізіотерапевтичні процедури. Без хірургічного втручання не обійтися при вродженій ваді серця, деформованому хребті. Пластична хірургія зменшує або ліквідує зовнішні ознаки.
Чи можна робити ЕКО?
Синдром Тернера супроводжується безплідністю. Можливість мати дітей є тільки у пацієнток з мозаїчною формою, у яких сформована матка. Статевий інфантилізм не дозволяє провести зачаття природним шляхом. Шанс виносити і народити здорову дитину є у жінки при проведенні ЕКО.
Якщо у жінки повноцінно розвинені яєчника, їй підсаджують власну запліднену яйцеклітину. Щоб дитина народилася без порушень на генному рівні, статеві клітини повинні мати нормальний каріотип 46ХХ. Сучасні репродуктивні методи дозволяють використовувати донорську яйцеклітину.
Успішне запліднення відбувається в 2-3 рази рідше, ніж у здорових жінок, і становить 20-30%. Ймовірність переривання вагітності після ЕКЗ складає у хворих пацієнток до 40%.
Профілактичні заходи
Хромосомна патологія діагностується у дітей здорових батьків. Передбачити можливість розвитку синдрому неможливо, як і вжити будь-яких заходів профілактики.
Єдиний спосіб виявити нестачу Х-хромосоми – пройти пренатальну діагностику на початкових термінах вагітності. На 11-13 тижні жінка може здати кров на наявність в ній плазменного протеїну А. Це означає підвищену ймовірність генетичної аномалії. Підтвердити або спростувати діагноз можна за допомогою біопсії ворсинок хоріона. За ним визначають каріотип плода, структуру хромосом.
Якщо синдром виявляється на стадії виношування, не потрібно панікувати. Дівчинка може народитися без зовнішніх проявів захворювання, а недорозвиненість статевих органів коригується з допомогою гормональної терапії.
